GIST "Contributing to Personalized Cancer Treatment"
 Photo by Asia Economy DB
Photo by Asia Economy DB
[Asia Economy Reporter Kim Bong-su] Domestic researchers have developed an algorithm that identifies genetic mutations in cancer cells and reconstructs their structure, promising to contribute to personalized treatment for cancer patients.
The research team led by Professor Lee Hyun-joo of the School of Electrical Engineering and Computer Science at Gwangju Institute of Science and Technology (GIST) announced on the 3rd that they developed a new graph-based algorithm that analyzes whole-genome sequencing data of cancer cells to detect genetic mutations and reconstruct genome structures at the single-nucleotide level.
The team succeeded in accurately detecting genetic mutations from whole-genome sequencing data?which often contains many detection errors?and identifying rearranged genome structures in cancer patients that were not discovered by existing methods. The human genome consists of 3 billion base pairs, and cancer cells have genetic mutations different from normal cells. Precisely identifying the distinct genetic mutations in each individual's cancer is crucial for personalized treatment.
The challenge lies in analyzing 3 billion base pairs to accurately identify mutations present in cancer cells, which is a very difficult task. In particular, the rearranged chromosome structures observed under microscopes in cancer cells have never been identified at the single-nucleotide level. Therefore, an algorithm capable of analyzing whole-genome data to detect these structures is needed.
The research team developed InfoGenomeR (Integrative Framework for Genome Reconstruction), a genetic mutation detection and genome reconstruction algorithm. It converts sequences with structural variations into graph forms and reconstructs the graph so that structural variations and copy number variations have consistent values, thereby reducing detection errors.
Subsequently, using heterozygous single nucleotide polymorphism information, they constructed a haplotype graph and restored the genome arrangement by finding an Eulerian path with the minimum entropy value.
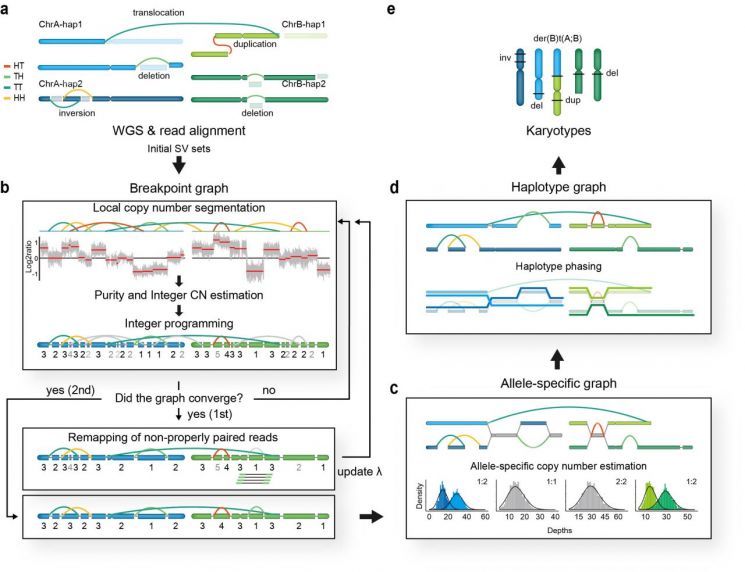
The InfoGenomeR developed by the research team significantly reduced genetic mutation detection errors (InfoGenomeR’s structural variation detection precision of 98.1% and F-measure of 94.9%) and reconstructed the genome arrangement of cancer cell lines at the single-nucleotide level. The accuracy of mutation detection was confirmed to be greatly improved compared to Manta, an algorithm by the international genome analysis company Illumina (Manta’s structural variation detection precision of 94.2% and F-measure of 90.4%).
The team applied the developed algorithm to breast and brain cancer patient data and identified that circular genome structures (double minutes) were amplified dozens of times in patients. This revealed that specific chromosomes undergo rearrangement processes depending on the cancer type. Additionally, they found that rearranged genomes newly appear in recurrent or metastatic cancers that were not present in the original cancer sites.
Professor Lee Hyun-joo said, “Reconstructing the genome arrangement of cancer cells at the single-nucleotide level using only whole-genome sequencing data is a challenging problem and was not possible with existing algorithms, but InfoGenomeR is the first algorithm to successfully accomplish this.” She added, “We hope to elucidate the regulation of cancer-related gene expression for personalized medicine based on the genome arrangement of cancer cells.”
The research results were published online on the 29th of last month in the international journal Nature Communications.
© The Asia Business Daily(www.asiae.co.kr). All rights reserved.
![Clutching a Stolen Dior Bag, Saying "I Hate Being Poor but Real"... The Grotesque Con of a "Human Knockoff" [Slate]](https://cwcontent.asiae.co.kr/asiaresize/183/2026021902243444107_1771435474.jpg)















{kind=link}