Kim Hyungbum's Team at Yonsei University College of Medicine
Comprehensive Functional Analysis of Over 27,000 ATM Gene Variants
A Korean research team has analyzed all variants of the ATM gene, which are associated with a high risk of cancer and closely linked to the development of rare diseases, and has elucidated their functions.

Severance Hospital announced on the 15th that Professor Kim Hyungbum, Lecturer Lee Kwangseop, and graduate student Min Jungoo from the Department of Pharmacology at Yonsei University College of Medicine have successfully evaluated all 27,513 single-nucleotide variants of the ATM gene, which is closely related to the development of cancer and rare diseases.
The ATM gene plays a crucial role in detecting and repairing DNA damage in the body. When this gene does not function properly, there is a significantly increased risk of developing cancers such as breast, colorectal, and pancreatic cancer, and the prognosis for cancer patients is often poor. It can also cause certain rare diseases, such as ataxia-telangiectasia. Therefore, identifying variants that impair the function of the ATM gene can help predict cancer risk in healthy individuals carrying these variants, as well as the treatment prognosis for cancer patients.
Although advances in genomic analysis technology have enabled more precise diagnoses of genetic diseases and cancer, many gene variants remain of unknown significance, making it difficult to use them appropriately in patient diagnosis and treatment. In particular, the ATM gene is a large gene with about 9,000 protein-coding amino acids and a high number of variants, making it difficult to evaluate using conventional statistical methods and thus challenging to apply in actual patient care and diagnosis.
The research team analyzed all 27,513 possible single-nucleotide variants in the entire protein-coding region (62 exons) of the ATM gene. Of these, 23,092 variants were directly assessed for function in cell experiments using the latest gene editing technology, prime editing, while the remaining 4,421 variants, which were difficult to evaluate experimentally, were assessed for their impact on cell viability using a self-developed artificial intelligence (AI) model called DeepATM.
As a result, the team was able to distinguish between variants that are harmful to the function of the ATM gene and those that are not, with high accuracy.
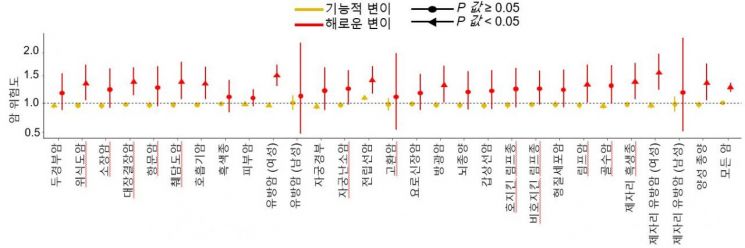 Using genomic and clinical data from approximately 500,000 individuals in the UK Biobank, the research team found that people carrying the harmful variants they identified have about a 1.4 times higher risk of developing cancer compared to those without such variants. Severance Hospital
Using genomic and clinical data from approximately 500,000 individuals in the UK Biobank, the research team found that people carrying the harmful variants they identified have about a 1.4 times higher risk of developing cancer compared to those without such variants. Severance Hospital
Subsequently, the research team used genomic and clinical data from approximately 500,000 individuals in the UK Biobank to validate their findings and confirmed that people carrying the harmful variants identified in the study have about a 1.4 times higher risk of developing cancer compared to those without such variants. They also demonstrated that their results matched more than 95% with data from ClinVar, an international genetic variant database. Furthermore, a reinterpretation of existing cancer genome data (cBioPortal) from the perspective of this study confirmed that cancer patient survival rates also differ depending on the presence of harmful ATM variants.
Professor Kim Hyungbum stated, "This study is significant in that it establishes the possibility of accurately distinguishing difficult-to-interpret ATM gene variants on a large scale," adding, "Similar analyses will become possible for other genes in the future, which will contribute to the advancement of genome-based precision medicine."
The results of this study were published in the latest issue of the international journal Cell (IF 42.5).
© The Asia Business Daily(www.asiae.co.kr). All rights reserved.
















{kind=link}
{kind=link}